We acquire and advance rare disease therapies that were deprioritized mid-development, ensuring they continue toward the patients who need them most.

Rugonersen
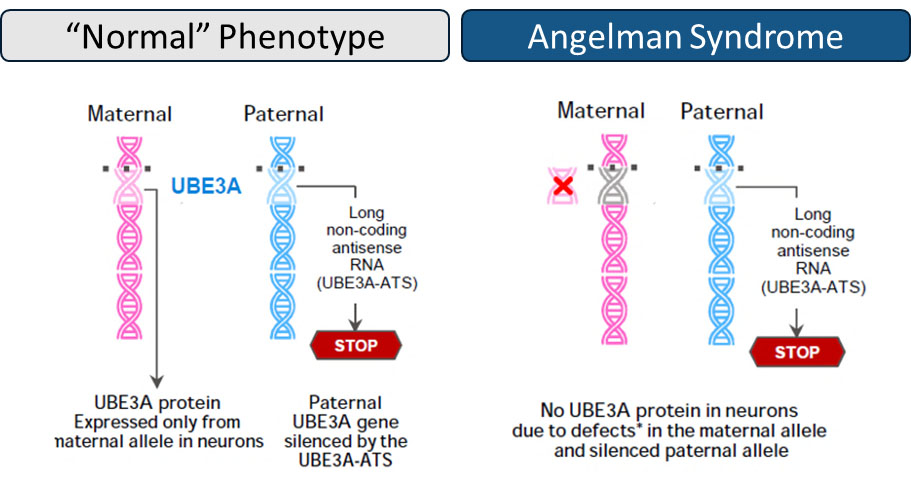
Angelman Syndrome (AS) is a serious rare genetic neurodevelopmental disorder which causes severe mental and physical impairment and affects approximately 15,000 patients in each of the US and the EU5, with an estimated incidence of 1 in 12,000 to 20,000 live births (Mertz et al., 2013; Luk and Lo, 2016; Yakoreva et al., 2019).
AS is characterized by global developmental delay, intellectual disability, epilepsy (90% of cases before age 3 years) with an atypical underlying electroencephalogram (EEG), ataxia, tremor, hyperactivity, limited speech, and sleep dysregulation (Albrecht et al. 1997; Kishino et al. 1997; Knoll et al. 1989; Thibert et al. 2013). Symptoms often emerge during infancy and persist throughout life (Albrecht et al. 1997; Kishino et al. 1997; Knoll et al. 1989). Natural history (NH) studies have shown that AS is also characterized by EEG abnormalities that are particularly pronounced in the lower frequency range (𝛿-frequency-range, defined here as 2-4 Hz). In AS, EEG 𝛿-power has been shown to correlate cross-sectionally and longitudinally with symptom severity (Frohlich, J. et al. 2019, Hipp, J. F. et al 2021, Ostrowski, L. M. et al. 2021). Affected individuals require lifelong care and cannot live independently.
Deletions and mutations in the maternal ubiquitin protein ligase E3A (UBE3A) allele cause AS, as paternal UBE3A is epigenetically silenced by a long non-coding antisense RNA (UBE3A-ATS) in neurons. UBE3A is required for normal brain development and function. Failure to express ubiquitin E3A ligase in central nervous system (CNS) neurons leads to a build-up of damaged or unwanted proteins, that if left unchecked, can paralyze normal neuronal maturation, function, and synaptic pruning.
Rugonersen, an ASO designed to address the underlying disease biology of Angelman syndrome (AS). Rugonersen specifically and potently binds the UBE3A-ATS transcript. Rugonersen binding triggers degradation of the UBE3A-ATS transcript in the CNS and therefore the unsilencing of the UBE3A paternal allele. Rugonersen allows neuronal expression of the paternal wild-type copy of the UBE3A gene, potentially restoring normal neuronal function and development in AS patients.
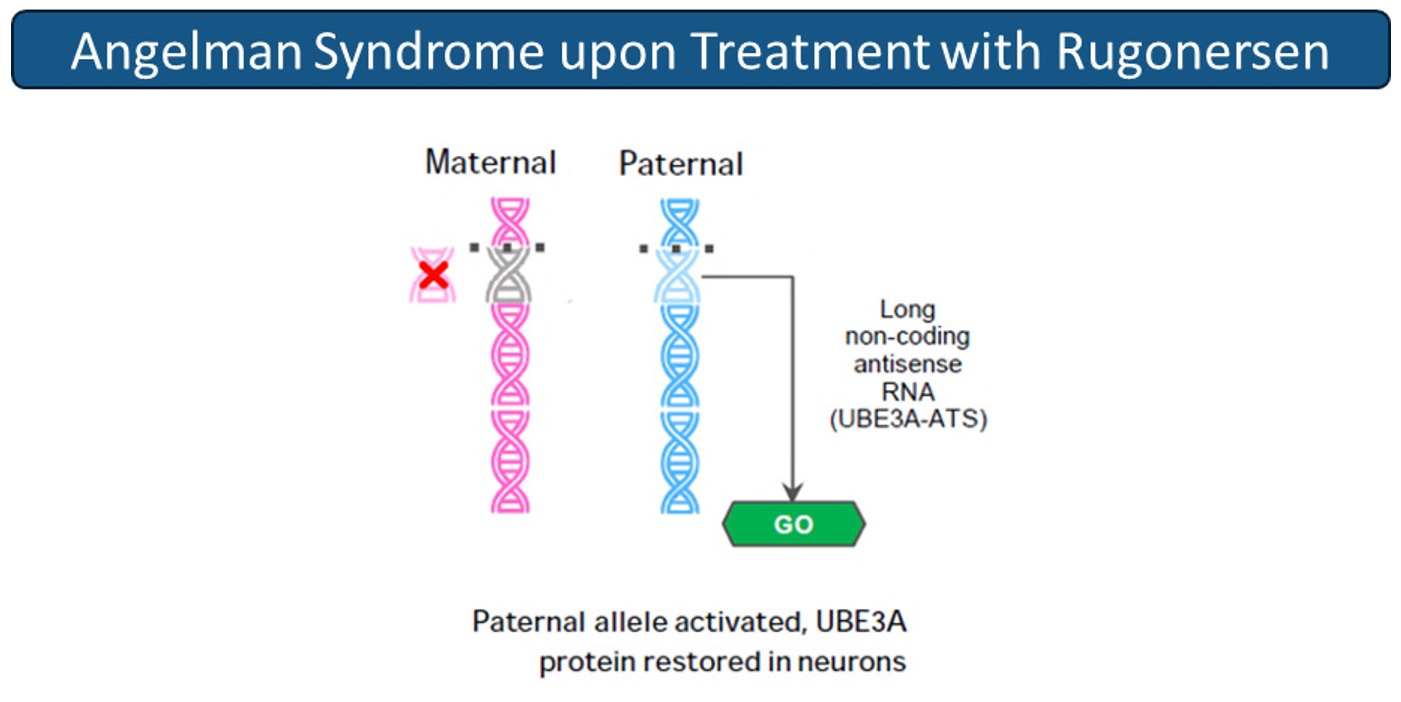
In nonclinical studies, rugonersen demonstrates the ability to reduce UBE3A-ATS and increase UBE3A mRNA and protein (Jagasia et al, 2025). In the TANGELO study, an open-label, non-randomized, adaptive study led by Roche, rugonersen demonstrated encouraging exploratory effects compared to natural history on multiple clinical measures and on a pharmacodynamic biomarker of brain function, EEG delta power.
The clinical data are published in detail in a recent publication in Nature Medicine: https://www.nature.com/articles/s41591-025-03784-7
On the basis of these promising results, OHB will be starting a Phase 3 study in the middle of 2026.
Hipp, J.F., Bacino, C.A., Bird, L.M. et al. The UBE3A-ATS antisense oligonucleotide rugonersen in children with Angelman syndrome: a phase 1 trial. Nat Med (2025). https://doi.org/10.1038/s41591-025-03784-7
Jagasia et al, Angelman syndrome patient-derived neuron screen leads to clinical ASO rugonersen targeting UBE3A-ATS with long-lasting effect in monkeys, Nucleic Acids Research (2025). https://doi.org/10.1093/nar/gkaf851